L'atrofia muscolare spinale (SMA) ha un'evoluzione diversa a seconda del bambino. L'età di insorgenza, i sintomi, le caratteristiche e la gravità della malattia differiscono notevolmente da un bambino all'altro.1
Le persone nelle immagini sono pazienti reali e, insieme alle loro famiglie, hanno fornito il consenso all’utilizzo delle loro storie. Le fotografie sono solo a scopo illustrativo.
Condividi la pagina
ATROFIA MUSCOLARE SPINALE NEI NEONATI E NEI BAMBINI
La SMA è una malattia neuromuscolare genetica rara1,2 e una delle principali cause genetiche di mortalità infantile.3
I bambini possono sperimentare una debolezza muscolare progressiva nei muscoli più vicini al centro del corpo, come spalle, cosce e bacino. Questi muscoli consentono attività come gattonare, camminare, sedersi e controllare il movimento della testa. Anche la capacità di respirare e deglutire può essere colpita.4
La SMA non colpisce i neuroni responsabili della cognizione, che è il processo mentale attraverso il quale acquisiamo conoscenza e comprensione mediante il pensiero, l'esperienza e i sensi.5,6
Secondo uno studio, i bambini e gli adolescenti con SMA hanno un'intelligenza normale, con un QI nell’intervallo standard.7
Condividi ora
LA SMA COLPISCE CIASCUN BAMBINO IN MANIERA DIVERSA
I sintomi possono includere debolezza muscolare progressiva, ipotonia e perdita di massa muscolare (atrofia). La debolezza muscolare colpisce in modo uguale entrambi i lati del corpo.9
Ciascun bambino può manifestare i sintomi in maniera differente e la malattia è classificata in diversi tipi in base all’età di insorgenza e alla capacità funzionale.
Condividi ora
Share now
IL MANCATO RAGGIUNGIMENTO DI ALCUNE TAPPE MOTORIE FONDAMENTALI PUÒ ESSERE IL PRIMO SEGNO DI SMA
Un genitore potrebbe sospettare che il proprio bambino sia affetto da atrofia muscolare spinale se non raggiunge alcune tappe fondamentali dello sviluppo motorio. I genitori potrebbero notare che il bambino non sta raggiungendo alcune tappe fondamentali dello sviluppo tipiche della sua età, come ad esempio la capacità di sostenere il capo, rotolarsi o sedersi autonomamente.
Anche la deglutizione o l’alimentazione possono diventare problematiche e i bambini possono perdere la capacità di deglutire in modo sicuro senza soffocare o inalare il cibo nei polmoni (aspirazione).5,10
Sebbene tutti i bambini si sviluppino secondo un proprio ritmo, l’Organizzazione Mondiale della Sanità (OMS) offre le seguenti linee guida delle tappe motorie quale parte dello studio Multicentre Growth Reference Study (MGRS).11
Share now
Adattato dal Multicentre Growth Reference Study dell’OMS.11
LINEE GUIDA SULLE TAPPE MOTORIE12
TAPPE MOTORIE FONDAMENTALI
CRITERI DI PRESTAZIONE
1.SEDERSI SENZA AIUTO
Il bambino si siede in posizione eretta con il capo eretto per ≥10 secondi. Non utilizza braccia o mani per bilanciare la posizione del corpo o del supporto.
2.GATTONARE SU MANI E GINOCCHIA
Il bambino si sposta in avanti o all’indietro su mani e ginocchia. Lo stomaco non tocca la superficie di appoggio. Movimenti continui e consecutivi, ≥3 di fila.
3.RESTARE IN POSIZIONE ERETTA CON ASSISTENZA
Il bambino resta in posizione eretta su entrambi i piedi, aggrappandosi a un oggetto stabile, come ad esempio un mobile. Il bambino rimane in posizione eretta con sostegno per ≥10 secondi.
4.DEAMBULARE CON ASSISTENZA
Il bambino si trova in posizione eretta con la schiena dritta. Effettua passi di lato o in avanti aggrappandosi a un oggetto stabile, con una o entrambe le mani. Una gamba si muove in avanti, mentre l’altra sostiene il peso corporeo. Il bambino effettua ≥5 passi.
5.RESTARE IN POSIZIONE ERETTA AUTONOMAMENTE
Il bambino sta in posizione eretta su entrambi i piedi (ma non sulle dita) con la schiena dritta. Le gambe sostengono il 100% del peso, senza alcun sostegno, per ≥10 secondi.
6.DEAMBULARE AUTONOMAMENTE
Il bambino è in grado di effettuare ≥5 passi da solo, con la schiena dritta. Una gamba si muove mentre l’altra sostiene la maggior parte del peso. Non vi è alcun contatto con persone o oggetti.
Condividi ora
CARATTERISTICHE DELLA SMA NEI NEONATI E NEI BAMBINI
Scopri di più sulle caratteristiche e sui diversi tipi di SMA a varie età di insorgenza:
Riesce a tenere la testa sollevata e inizia a spingere verso l'alto quando è sdraiato a pancia in giù; effettua movimenti più fluidi con braccia e gambe.
4 mesi:
Tiene la testa ferma, senza sostegno; spinge verso il basso le gambe quando i piedi sono su una superficie dura; può essere in grado di rotolare dalla pancia alla schiena; può tenere in mano un giocattolo, scuoterlo e far oscillare giocattoli pendenti; porta le mani alla bocca; quando è sdraiato sulla pancia, spinge sui gomiti.
6 mesi:
Si rotola in entrambe le direzioni (da davanti a dietro e da dietro a davanti); inizia a sedersi senza sostegno; quando è in piedi, sostiene il peso sulle gambe e potrebbe eseguire dei balzi; si dondola avanti e indietro, a volte strisciando all'indietro prima di andare avanti.
Massima tappa motoria fondamentale raggiunta
Aspettativa di vita
Tipo di SMA
Massima tappa motoria fondamentale raggiunta
Non è in grado di stare seduto (“non-sitter”)
Aspettativa di vita
≤ 2 anni13
Tipo di SMA
Tipo 1 (noto anche come malattia di Werdnig-Hoffmann)
Caratteristiche:13,16,17
Scarso controllo della testa
Tosse debole
Pianto debole
Debolezza progressiva dei muscoli utilizzati per masticare e deglutire
Scarso tono muscolare
Postura “a gamba di rana” quando è sdraiato
Grave debolezza muscolare su entrambi i lati del corpo
Debolezza progressiva dei muscoli che aiutano la respirazione (muscoli intercostali), con il risultato del caratteristico torace “a campana”
Share now
TAPPE MOTORIE DI SVILUPPO:10
9 mesi:
Sta in piedi, aggrappandosi ad un sostegno; può mettersi in posizione seduta; sta seduto senza supporto; si tira su per stare in piedi; gattona.
1 anno:
Si mette in posizione seduta senza aiuto; si tira su per stare in piedi, cammina aggrappandosi ai mobili (cruising); può fare qualche passo senza aggrapparsi; può stare in piedi da solo.
Massima tappa motoria fondamentale raggiunta
Aspettativa di vita
Tipo di SMA
Massima tappa motoria fondamentale raggiunta
È in grado di stare seduto in modo autonomo (“sitter”)
Aspettativa di vita
> 2 anni13 Il 70% è ancora vivo all'età di 25 anni
Tipo di SMA
Tipo 2 (anche nota come malattia di Dubowitz)
Caratteristiche:16,17
Debolezza muscolare
Possono verificarsi problemi di deglutizione, tosse e respirazione, ma in genere sono meno comuni
Dolori muscolari e sintomi di rigidità articolare
I bambini possono sviluppare problemi alla colonna vertebrale, come la scoliosi (curvatura della colonna vertebrale), che possono richiedere l’utilizzo di un tutore o di un intervento chirurgico
Share now
Massima tappa motoria fondamentale raggiunta
Aspettativa di vita
Tipo di SMA
Massima tappa motoria fondamentale raggiunta
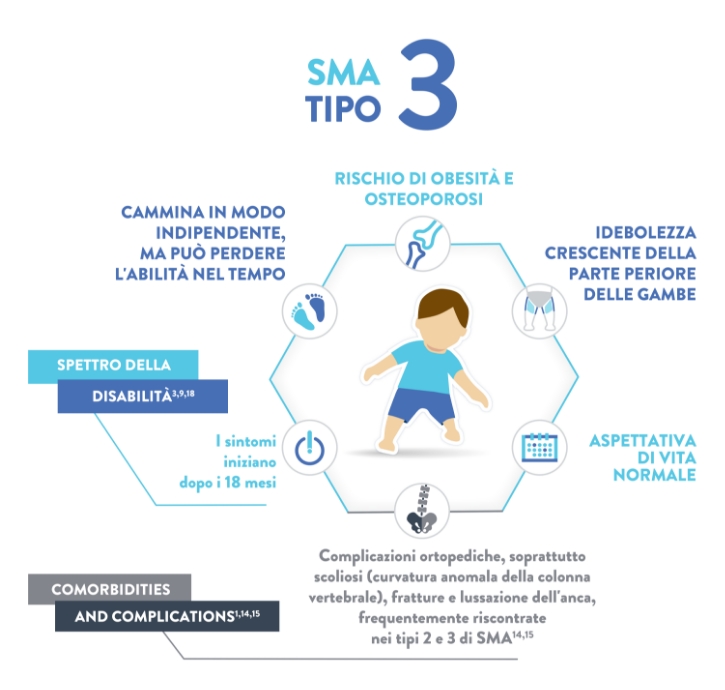
È in grado di camminare in modo autonomo (“walker” – anche se può progressivamente perdere questa capacità)
Aspettativa di vita
Normale13
Tipo di SMA
Tipo 3 (anche nota come malattia di Kugelberg-Welander)
Caratteristiche:16,17
Scoliosi
Difficoltà di masticazione e deglutizione
I muscoli delle gambe sono generalmente più colpiti di quelli delle braccia
Debolezza progressiva dei muscoli utilizzati per masticare e deglutire
Dolori muscolari
Sintomi da uso eccessivo delle articolazioni
Share now
Massima tappa motoria fondamentale raggiunta
Aspettativa di vita
Tipo di SMA
Massima tappa motoria fondamentale raggiunta
Tutte
Aspettativa di vita
Normale13
Tipo di SMA
Tipo 4
Caratteristiche:16,17
I sintomi fisici sono simili a quelli dell'atrofia muscolare spinale a esordio giovanile, con insorgenza graduale di debolezza, tremori e contrazioni muscolari, osservati per la prima volta nella tarda adolescenza o nella prima età adulta.
Share now
Condividi ora
I resoconti dei genitori sullo sviluppo motorio dei figli tendono ad essere affidabili. Condividere le osservazioni di potenziali ritardi motori con un medico può aiutare a determinare una strategia di cura appropriata.18,19
Scopri quali sono i campanelli d'allarme che potrebbero farti sospettare che un individuo abbia la SMA.
Le persone nelle immagini sono pazienti reali e, insieme alle loro famiglie, hanno fornito il consenso all’utilizzo delle loro storie. Le fotografie sono solo a scopo illustrativo.
RIFERIMENTI
1. Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.
3. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
4. Finkel R, et al. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9. November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593-602.
5. Qian Y., McGraw S., Henne J., Jarecki J., Hobby K., Yeh W.S. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: A qualitative study. BMC Neurol. 2015;15:1–12. doi: 10.1186/s12883-015-0473-3.
7. Von Gontard et al. Intelligence and cognitive function in children and adolescents with spinal muscular atrophy. Neuromuscul Disord. 2002. Feb;12(2):130-6.
9. D’Amico A, et al. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71.
10. Centers for Disease Control and Prevention. Developmental milestones. Available at: http://www.cdc.gov/ncbddd/actearly/milestones/. Updated January 21, 2016. Accessed April 27, 2016.
12. Wijnhoven TMA, de Onis M, Onyango AE, et al; for the WHO Multicentre Growth Reference Study Group. Assessment of gross motor development in the WHO Multricentre Growth Reference Study. Food Nutr Bull. 2004;25(1 suppl 1):S37-S45.
13. Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46(1):1-12.
14. Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet 2013;6:113-120.
15. Darras BT. Spinal muscular atrophies. Paediatr Clin North Am 2015;62(3):743-766. DOI: 10.1016/j.pcl.2015.03.010.
16. Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscl Disord. 2018;28(2):103-115.
17. Prior TW, Russman BS. Spinal muscular atrophy. NCBI Bookshelf Website. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1352/. Updated November 14, 2013. Accessed April 15, 2016.
19. Noritz GH, Murphy NA; and Neuromuscular Screening Expert Panel. Motor delays: early identification and evaluation. Pediatrics. 2013;131(6):e2016-e2027.
20. Lawton S, Hickerton C, Archibald AD, McClaren BJ, Metcalfe SA. A mixed methods exploration of families’ experiences of the diagnosis of childhood spinal muscular atrophy. Eur J Hum Genet. 2015;23(5):575-580.